
Vidjil is an open-source platform for the analysis of high-throughput sequencing data from lymphocytes, developed by the Bonsai bioinformatics lab and the VidjilNet consortium.
V(D)J recombinations in lymphocytes are essential for immunological diversity. They are also useful markers of pathologies, and in leukemia, are used to quantify the minimal residual disease during patient follow-up. High-throughput sequencing (NGS/HTS) now enables the deep sequencing of a lymphoid population with dedicated sequencing methods and software, called either Rep-Seq or AIRR-Seq.
Vidjil is used in routine clinical practice in hospitals around the world, in particular for the diagnosis of patients suffering Acute Lymphoblastic Leukemia (ALL). More than 10,000 samples of ALL patients were analyzed with Vidjil since 2015. Vidjil is also used in 40+ studies on hemopathies (ALL, CLL, lymphomas, WM...) and immunology topics involving T-cell or B-cell repertoires. Vidjil was awarded an honourable mention at the 2022 Open Science Awards for Open Source Research Software, in the Community category.
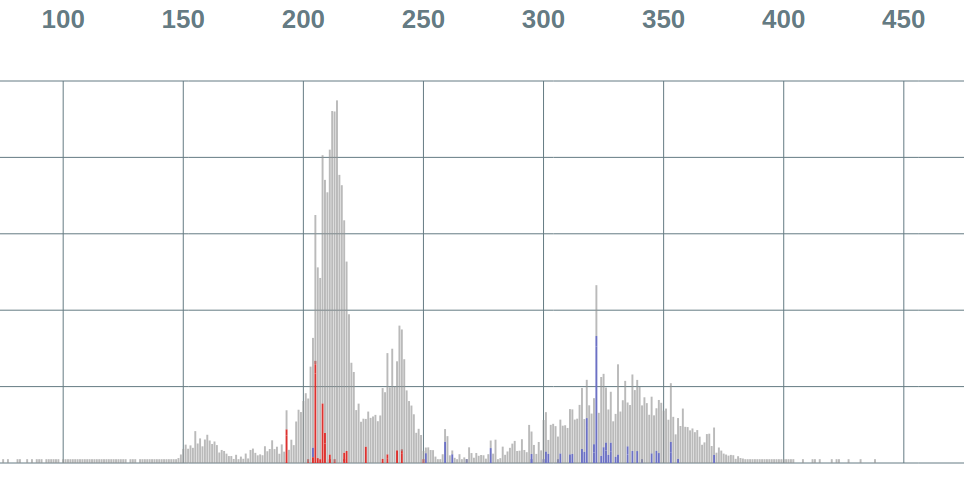
Repertoire analysis. Vidjil analyses the entire B/T immune repertoire. The Genescan-like view, here colored by locus, gives a first approach of the diversity of the sample and allows to assess the mono/poly-clonality.
![]()
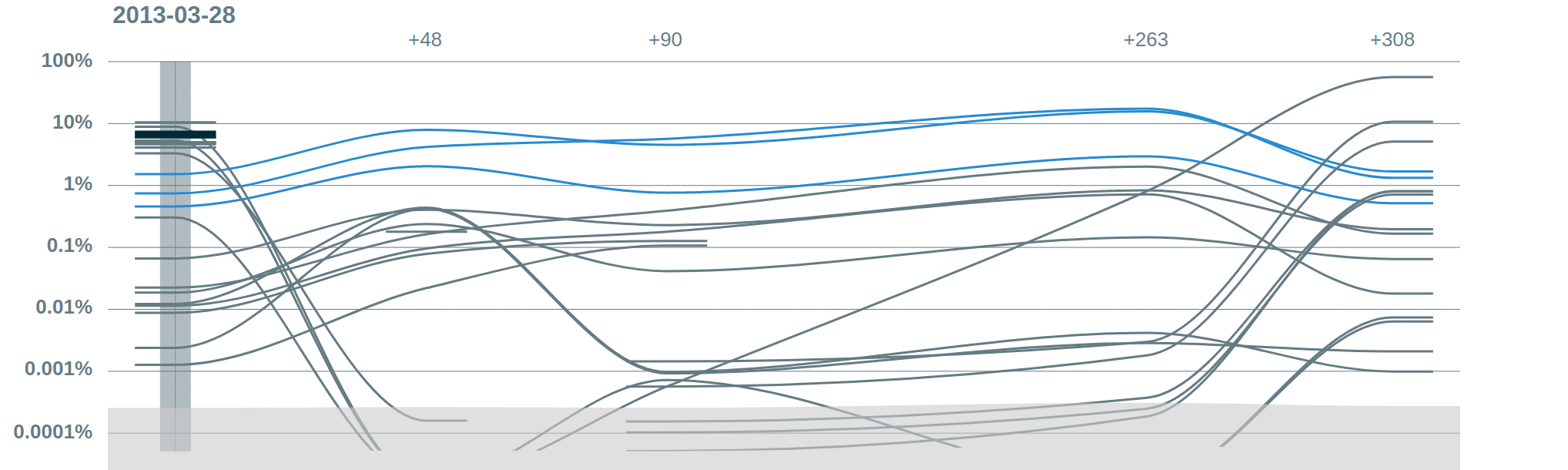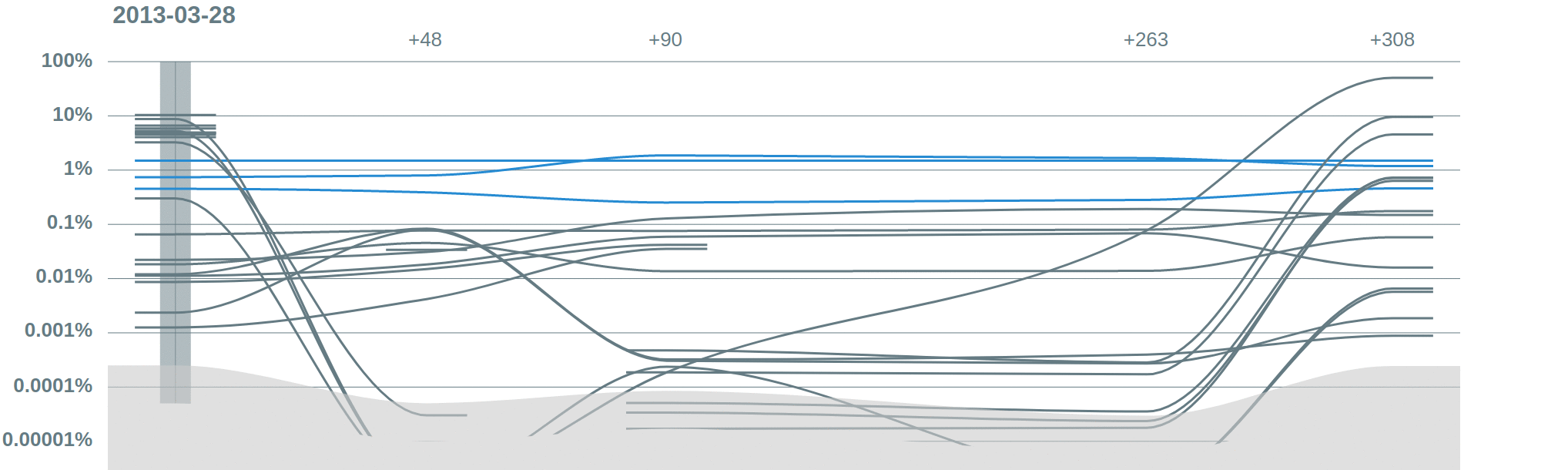
Clone tracking. Vidjil allows to compare several samples and to track clones along the time. This ALL patient has a relapse at day 308 on (minor) clones that were present at the diagnosis and that expanded troughout time.
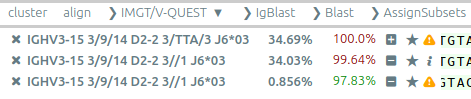
CLL, productivity analysis. For CLL as well as immunological analyses, Vidjil estimates the productivity of each clone. It can also display information computed by IMGT/V-QUEST, such as on hypermutations.
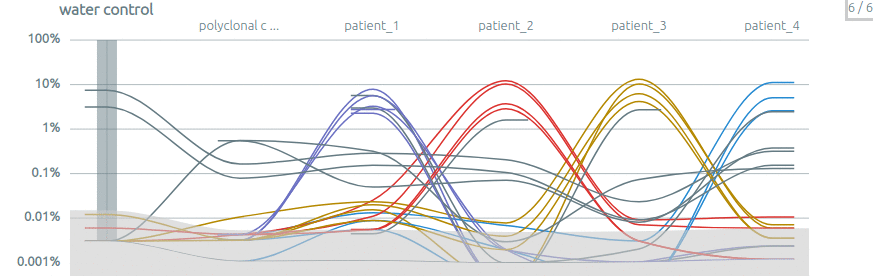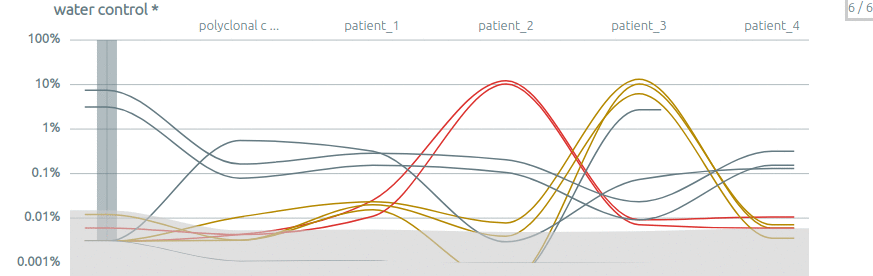
Intra-run contamination. The comparison between several samples can be used to assess the inter-sample contamination, notably when there is a control sample.
Spikes. Vidjil offer several normalization options, for example where there is a control spike with known abundance.

At the heart of the Vidjil platform, Vidjil-algo processes high-througput sequencing data to extract V(D)J junctions and gather them into clones. Vidjil-algo starts from a set of reads and detects “windows” overlapping the actual CDR3. It detects gene rearrangements from both immunoglobulins and T-cell receptors, as well as some incomplete or uncommon rearrangements. The analysis is based on a reliable seed-based algorithm. It is extremely fast because, in the first phase, no alignment is performed with database germline sequences. The algorithm works on reads coming from either amplicon-based or capture-based deep sequencing strategy, as soon as they include CDR3 sequences. Both human and mouse immune systems can be analyzed.

The Vijdil web application is made for the visualization, inspection and analysis of clones and their tracking along the time in a MRD setup or in a immunological study. The application can visualize data processed by the Vidjil algorithm or by other V(D)J analysis pipelines. It enables to explore further clusterings proposed by the software and/or done manually done by the user.

The web application can be linked to a patient/experiment and sample database. After authentication, the clinicians or researchers upload .fasta/.fastq/.gz files, manage and process their runs directly from the web application. They can save their analysis and generate reports for the patient record. The server with the sample database can be installed in any hospital or computer center. A public web server is also available since October 2014. More than 20 labs in Europe and in the world regularly use Vidjil through the web application. In 18 months, the public server analyzed more than 5,000,000 sequences from 5,000 AIRR/RepSeq samples.

Strong attention to community, users, and stability. High-quality development process, with systemating testing and continous integration.
We welcome new members in the VidjilNet consortium. We also welcome Bitcoin donations to 13u12m6LxVhesKEpS6T5wpYN19LHpwk8xt. Thank you for your support !
All the code of Vidjil is available under open-source licenses (GPLv3, as well as some other free licences for some third-parties librairies). We also offer extended support as well as custom development for various types of projects. Please contact us if you are interested.
Vidjil is developed by a passionate team with fast response, from the VidjilNet consortium and the Bonsai bioinformatics team of the CRIStAL (CNRS, U. Lille) and Inria Lille research centers, in Lille, France. This work is in collaboration with the department of Hematology of CHRU Lille, the Functional and Structural Genomic Platform (U. Lille 2, IFR-114, IRCL), and the EuroClonality-NGS working group, and is supported by SIRIC ONCOLille (Grant INCa-DGOS-Inserm 6041), Région Nord-Pas-de-Calais (ABILES), Université Lille (the scientific computing service, Mésocentre de Lille for hosting our servers, as well as the former PPF bioinformatique), and Inria Lille. The methods were presented at the JOBIM 2013 and ASH 2014 conferences, and are described in the following papers:
Chrystelle Abdo et al., Caution encouraged in next-generation sequencing immunogenetic analyses in acute lymphoblastic leukemia, Blood, 2020, 136(9):1105–1107, https://doi.org/10.1182/blood.2020005613
Jean-Sebastien Allain et al., IGHV segment utilization in immunoglobulin gene rearrangement differentiates patients with anti-myelin-associated glycoprotein neuropathy from others immunoglobulin M-gammopathies, Haematologica, 2018, 103:e207-e210, http://dx.doi.org/10.3324/haematol.2017.177444
Kristian Assing et al., A Novel CDC42 Variant with Impaired Thymopoiesis, IL-7R Signaling, PAK1 Binding, and TCR Repertoire Diversity Journal of Clinical Immunology, 2023, https://doi.org/10.1007/s10875-023-01561-0
Jack Bartram et al., High throughput sequencing in acute lymphoblastic leukemia reveals clonal architecture of central nervous system and bone marrow compartments, Haematologica, 2018, https://dx.doi.org/10.3324%2Fhaematol.2017.174987
Sébastien Bender et al., Immunoglobulin variable domain high-throughput sequencing reveals specific novel mutational patterns in POEMS syndrome, Blood, 2020, https://doi.org/10.1182/blood.2019004197
Marie-Laure Boulland et al., Reliable IGHV status assessment by next generation sequencing in routine practice for chronic lymphocytic leukemia, Leukemia & Lymphoma, 2021, https://doi.org/10.1080/10428194.2021.1933476
Estelle Bourbon et al., Next-CLL: A New Next-Generation Sequencing–Based Method for Assessment of IGHV Gene Mutational Status in Chronic Lymphoid Leukemia, The Journal of Molecular Diagnostics, 2023, https://doi.org/10.1016/j.jmoldx.2023.01.009
Monika Brüggemann et al., on behalf of the EuroClonality-NGS working group, Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study, Leukemia, 2019, 33, 2241–2253, https://doi.org/10.1038/s41375-019-0496-7
Roberta Cavagna et al., Capture-based Next-Generation Sequencing Improves the Identification of Immunoglobulin/T-Cell Receptor Clonal Markers and Gene Mutations in Adult Acute Lymphoblastic Leukemia Patients Lacking Molecular Probes, Cancers, 2020, 12(6), 1505, https://doi.org/10.3390/cancers12061505
Rodolfo P. Correia et al., High‐throughput sequencing of immunoglobulin heavy chain for minimal residual disease detection in B‐lymphoblastic leukemia, Int. Journal of Laboratory Hematology, 2021, https://doi.org/10.1111/ijlh.13453
Frédéric Davi et al., on behalf of ERIC, the European Research Initiative on CLL, and the EuroClonality-NGS Working Group, Immunoglobulin gene analysis in chronic lymphocytic leukemia in the era of next generation sequencing, 2020 Leukemia, 2020, https://doi.org/10.1038/s41375-020-0923-9
Rachel Dobson et al., Widespread in situ follicular neoplasia in patients who subsequently developed follicular lymphoma, The Journal of Pathology, 2021, https://doi.org/10.1002/path.5861
Yann Ferret et al., Multi-loci diagnosis of acute lymphoblastic leukaemia with high-throughput sequencing and bioinformatics analysis, British Journal of Haematology, 2016, 173, 413–420, https://hal.archives-ouvertes.fr/hal-01279160
Henrike J. Fischer et al., Modulation of CNS autoimmune responses by CD8+ T cells coincides with their oligoclonal expansion, Journal of Neuroimmunology, 2015, S0165-5728(15)30065-5, http://dx.doi.org/10.1016/j.jneuroim.2015.10.020
Navarro Nilo Giusti et al., Test trial of spike-in immunoglobulin heavy-chain (IGH) controls for next generation sequencing quantification of minimal residual disease in acute lymphoblastic leukaemia, British Journal of Haematology, 2020, 189: e150-e154, https://doi.org/10.1111/bjh.16571
Isadoral Heraud et al., Monoclonal B-cell lymphocytosis with a non-CLL immunophenotype–Review of 34 cases Annales de Biologie Clinique, 2023, https://www.jle.com./fr/revues/abc/e-docs/monoclonal_b_cell_lymphocytosis_with_a_non_cll_immunophenotype_review_of_34_cases_330642/article.phtml?tab=texte
Irene Jo et al., Considerations for monitoring minimal residual disease using immunoglobulin clonality in patients with precursor B-cell lymphoblastic leukemia, Clinica Chimica Acta, 2019, https://doi.org/10.1016/j.cca.2018.10.037
Natalia Izotova et al., Long-term lymphoid progenitors independently sustain naïve T and NK cell production in humans, Nature Communications, 2021, https://doi.org/10.1038/s41467-021-21834-9
Vincent Jauvague et al., RNA-based immunoglobulin repertoire sequencing is a new tool for the management of monoclonal gammopathy of renal (kidney) significance. Kidney International, 2022, 101(2), 331-337, https://doi.org/10.1016/j.kint.2021.10.017
Takashi Kanamori et al., Genomic analysis of multiple myeloma using targeted capture sequencing in the Japanese cohort, British Journal of Haematology, 2020, https://doi.org/10.1111/bjh.16720
Kenji Kimura et al., Identification of Clonal Immunoglobulin λ Light-Chain Gene Rearrangements in AL Amyloidosis Using Next-Generation Sequencing, Experimental Hematology, 2021, 101:34-41.e4 https://doi.org/10.1016/j.exphem.2021.08.001
Michaela Kotrova et al., The predictive strength of next-generation sequencing MRD detection for relapse compared with current methods in childhood ALL, Blood, 2015, 126:1045-1047, http://dx.doi.org/10.1182/blood-2015-07-655159
Michaela Kotrova et al., Next‐generation amplicon TRB locus sequencing can overcome limitations of flow‐cytometric Vβ expression analysis and confirms clonality in all T‐cell ,prolymphocytic leukemia cases, Cytometry Part A, 93(11):1118-1124, 2018, http://dx.doi.org/10.1002/cyto.a.23604
Anton W. Langerak, High-Throughput Immunogenetics for Clinical and Research Applications in Immunohematology: Potential and Challenges, Journal of Immunology, 2017, 198(10):3765-3774, https://dx.doi.org/10.4049/jimmunol.1602050
Yannick Le Bris et al., Single Capture High Throughput Sequencing Assay for Combined V(D)J Clonality Analysis and Oncogene Mutations in the Diagnosis of T and B Lymphoid Malignancies, ASH 2021, Blood, 138(S1):2404, https://doi.org/10.1182/blood-2021-151083
Zhenhua Li et al., Identifying IGH disease clones for MRD monitoring in childhood B-cell acute lymphoblastic leukemia using RNA-Seq, Leukemia, 2020, 34:2418-2429, http://dx.doi.org/10.1038/s41375-020-0774-4
Ralf A. Linker et al., Thymocyte-derived BDNF influences T-cell maturation at the DN3/DN4 transition stage, European Journal of Immunology, 2015, 45, 1326-1338, http://dx.doi.org/10.1002/eji.201444985
Ming Liang Oon et al., T-Cell Lymphoma Clonality by Copy Number Variation Analysis of T-Cell Receptor Genes, Cancers, 2021, 13(2), 340, https://dx.doi.org/10.3390/cancers13020340
Alejandro Medina et al., Comparison of next-generation sequencing (NGS) and next-generation flow (NGF) for minimal residual disease (MRD) assessment in multiple myeloma, Blood Cancer Journal, 10, 108, 2020, https://doi.org/10.1038/s41408-020-00377-0
Dai Nishijima et al., Capture Sequencing Is a Useful Method for Comprehensive Clonality Analysis Based on Ig/TCR Gene Rearrangements in Acute Lymphoblastic Leukemia, ASH 2018, Blood, 132(S1):1543, https://doi.org/10.1182/blood-2018-99-115624
Alexis Piedrafita et al., Spectrum of Kidney Disorders Associated with T-Cell Immunoclones, Journal of Clinical Medicine, 2022, 11(3), 604, https://doi.org/10.3390/jcm11030604
Edit Porpaczy et al., Aggressive B-cell lymphomas in patients with myelofibrosis receiving JAK1/2 inhibitor therapy, Blood, 2018, https://dx.doi.org/10.1182/blood-2017-10-810739
Natalia Izotova et al., Long-term lymphoid progenitors independently sustain naïve T and NK cell production in humans, Nature Communications, 2021, 12:1622, https://doi.org/10.1038/s41467-021-21834-9
Rathana Kim et al., Adult T-cell acute lymphoblastic leukemias with IL7R pathway mutations are slow-responders who do not benefit from allogeneic stem-cell transplantation, Leukemia, 2020, 34, 1730-1740, https://dx.doi.org/10.1038/s41375-019-0685-4
Cédric Pastoret et al., Molecular mechanisms underlying transformation of large granular lymphocytic leukemia to high-grade T-cell lymphoma, Leukemia, 2023, https://www.nature.com/articles/s41375-023-01922-z
Mikaël Salson et al., High-throughput sequencing in acute lymphoblastic leukemia: Follow-up of minimal residual disease and emergence of new clones, Leukemia Research, 2017, 53, 1–7, http://dx.doi.org/10.1016/j.leukres.2016.11.009
Masashi Sanada et al., Targeted-Capture Sequencing Is a Useful Method for MRD Markers Screening in KMT2A (MLL) Rearranged Leukemia, ASH 2019, Blood, 134(S1):2759, https://doi.org/10.1182/blood-2019-125421
Florian Scherer et al., Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA, Science Translational Medicine, 2016, 8, 364ra155, http://dx.doi.org/10.1126/scitranslmed.aai8545
V. Seitz et al., Evidence for a role of RUNX1 as recombinase cofactor for TCRβ rearrangements and pathological deletions in ETV6-RUNX1 ALL Scientific Reports, 2020, 10:10024, https://doi.org/10.1038/s41598-020-65744-0
Udo zur Stadt et al., Characterization of novel, recurrent genomic rearrangements as sensitive MRD targets in childhood B-cell precursor ALL, Blood Cancer Journal, 2019, https://doi.org/10.1038/s41408-019-0257-x
Lucia Stranavova et al., Heterologous Cytomegalovirus and Allo-Reactivity by Shared T Cell Receptor Repertoire in Kidney Transplantation, Frontiers in Immunology, 2019, https://doi.org/10.3389/fimmu.2019.02549
Manuela Tosi et al., MRD-Based Therapeutic Decisions in Genetically Defined Subsets of Adolescents and Young Adult Philadelphia-Negative ALL Cancers 2021, 13(9), 2108, https://doi.org/10.3390/cancers13092108
Amelie Trinquand et al., Toward Pediatric T Lymphoblastic Lymphoma Stratification Based on Minimal Disseminated Disease and NOTCH1/FBXW7 Status, HemaSphere, 2021, https://doi.org/10.1097/hs9.0000000000000641
Christine Wennerås et al., Infection with Neoehrlichia mikurensis promotes the development of malignant B-cell lymphomas , British Journal of Haematology, 2023, https://doi.org/10.1111/bjh.18652
Gary Wright et al., Clinical benefit of a high‐throughput sequencing approach for minimal residual disease in acute lymphoblastic leukemia, Pediatric Blood & Cancer, 2019, https://doi.org/10.1002/pbc.27787
Wen‐Qing Yao et al., Angioimmunoblastic T‐cell lymphoma contains multiple clonal T‐cell populations derived from a common TET2 mutant progenitor cell, The Journal of Pathology, 2019, https://doi.org/10.1002/path.5376
Yasuda et al., Clinical utility of target capture‐based panel sequencing in hematological malignancies: A multicenter feasibility study, Cancer Science, 2020, 111(9):3367-3378, https://dx.doi.org/10.1111/cas.14552